|
| |
|
|
 |
|
Une enzyme cruciale enfin démasquée |
|
|
| |
|
| |
Une enzyme cruciale enfin démasquée
20 NOV 2017 | PAR INSERM (SALLE DE PRESSE) | BIOLOGIE CELLULAIRE, DÉVELOPPEMENT ET ÉVOLUTION | CANCER | NEUROSCIENCES, SCIENCES COGNITIVES, NEUROLOGIE, PSYCHIATRIE
Après 40 ans de recherche, des chercheurs du CEA, du CNRS, de l’Université Grenoble Alpes, de l’Université de Montpellier et de l’Inserm ont enfin démasqué l’enzyme responsable de la détyrosination de la tubuline. Surprise : ce n’est pas une enzyme mais deux qui ont été découvertes capables de modifier ce composant essentiel du squelette de la cellule. Ces travaux ouvrent de nouvelles pistes pour mieux comprendre le rôle de la tubuline dont les altérations accompagnent cancers, maladies cardiaques et défauts neuronaux. Ces résultats sont publiés le 16 novembre 2017 dans la revue Science.
Une collaboration internationale impliquant des chercheurs du CEA, du CNRS, de l’Inserm, de l’Université Grenoble Alpes, de l’Université de Montpellier et de l’Université de Stanford[1] a identifié une enzyme, la Tubuline CarboxyPeptidase (TCP), qui est responsable d’une transformation biochimique des microtubules cellulaires, la détyrosination. La détyrosination est une réaction biologique consistant à supprimer l’acide aminé terminal tyrosine[2], de la tubuline α, un composant des microtubules. Alors qu’elle était recherchée depuis quatre décennies, les biologistes ont réussi à isoler cette protéine par purification et ont ensuite apporté les preuves de son activité cellulaire.
Les microtubules contribuent à des fonctions cellulaires essentielles
Les microtubules sont des fibres dynamiques présentes dans toutes les cellules. Formés par l’assemblage de deux protéines (tubuline α et tubuline β), les microtubules assurent de nombreuses fonctions. Ils séparent les chromosomes destinés aux deux cellules filles lors de la division cellulaire, ils contribuent à la polarité des cellules, à la morphologie et à la migration cellulaire. Ils forment des sortes de rails sur lesquels sont transportés des constituants cellulaires tels que des protéines ou des brins d’ARN.
Ces fonctions cellulaires sont régulées grâce à l’existence de « signaux » présents à la surface des microtubules. Ces signaux sont des modifications biochimiques des acides aminés (appelées modifications post-traductionnelles car elles ont lieu après la synthèse de la protéine) qui sont réalisées par plusieurs enzymes qui, ici, modifient les tubulines.
L’enzyme TCP, identifiée après 40 ans de mystère
L’activité de l’une de ces enzymes a été mise en évidence pour la première fois en 1977 par des chercheurs argentins qui lui donnent le nom de TCP (Tubuline CarboxyPeptidase). Cette enzyme, qui n’avait jusqu’à ce jour jamais été identifiée (sa taille et sa séquence restaient inconnues), a comme fonction de supprimer le dernier acide aminé, une tyrosine, de l’extrémité de la tubuline α. C’est la réaction de détyrosination. Une enzyme réverse, la ligase TTL, est chargée de repositionner cette tyrosine à sa place. C’est la tyrosination. Ce cycle de détyrosination/tyrosination est vital pour la cellule et l’organisme. Une détyrosination massive (anormale) est observée dans plusieurs cancers sévères et maladies cardiaques.
Identifier et caractériser la TCP constituait donc un objectif majeur pour comprendre la fonction physiologique de la détyrosination de la tubuline α et pour évaluer les conséquences de son inhibition.
Pour isoler la TCP, les chercheurs ont suivi son activité, utilisé des techniques classiques de biochimie et fait appel à des chimistes de l’Université de Stanford qui ont développé une petite molécule inhibitrice de son activité. Cette molécule a été utilisée comme hameçon pour « pêcher » l’enzyme convoitée.
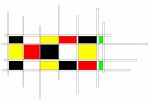
Cycle de détyrosination/tyrosination de la tubuline
Les microtubules sont des fibres présentes dans toutes les cellules composées d’un empilement de tubulines α/β. La tubuline α porte une tyrosine (Y) à son extrémité qui est alternativement enlevée et replacée par deux enzymes, modifiant ainsi la surface des microtubules. La TCP (représentée par une scie composée de deux éléments, VASH/SVBP) est responsable de la détyrosination. La TTL (représentée par un tube de colle) replace la tyrosine sur la tubuline. Ce cycle est essentiel aux diverses fonctions des microtubules dans les cellules (division, migration, …) et vital pour l’organisme. © C. Bosc, GIN
Au final, ce ne sont pas une, mais deux enzymes qui ont été découvertes ! Ces dernières, dénommées VASH1 et VASH2, étaient déjà connues des scientifiques mais sans savoir qu’il s’agissait d’enzymes en lien avec le cytosquelette. Les chercheurs ont montré qu’à la condition d’être associées à une protéine partenaire appelée SVBP, VASH1 et VASH2 sont capables de détyrosiner la tubuline α. Pour le démontrer, les chercheurs ont supprimé leur expression (ou celle de leur partenaire SVBP) dans les neurones. Ils ont alors observé une très forte diminution du taux de détyrosination de la tubuline α, ainsi que des anomalies dans la morphologie des neurones (v. Figure). Les chercheurs sont allés plus loin en montrant que ces enzymes sont également impliquées dans le développement du cortex cérébral.
Des perspectives pour la lutte contre le cancer
Ainsi, quarante ans après les premiers travaux sur la détyrosination de la tubuline α, les enzymes responsables ont été démasquées ! Dorénavant, les scientifiques espèrent qu’en modulant l’efficacité de la TCP et en améliorant les connaissances du cycle détyrosination/tyrosination, ils pourront mieux lutter contre certains cancers et progresseront dans la connaissance des fonctions cérébrales et cardiaques.
Photographies de l’altération des neurones par une réduction de l’expression des enzymes TCP (VASH/SVBP. De gauche à droite : neurone contrôle, neurones dans lesquels l’expression de VASH1 et VASH2 est réduite, neurones dans lesquels l’expression de SVBP est réduite. Les neurones ayant moins d’enzyme présentent un retard de développement et des anomalies morphologiques. © L. Peris /GIN
[1] Les instituts suivants sont impliqués : Grenoble Institut des neurosciences, GIN (Inserm/Univ. Grenoble Alpes); l’Institut de biosciences et biotechnologies de Grenoble, BIG (Inserm/CEA/Univ. Grenoble Alpes) ; l’Institut pour l’avancée des biosciences, IAB (Inserm/CNRS/Univ. Grenoble Alpes), le Department of Pathology, Stanford University School of Medicine (Stanford, USA), l’Institut de génétique humaine, IGH (CNRS/Univ. de Montpellier), le Centre de recherche en biologie cellulaire de Montpellier, CRBM (CNRS/Univ. de Montpellier).
[2] La tyrosine est l’un des 22 acides aminés qui constituent les protéines
DOCUMENT inserm LIEN |
| |
|
| |
|
|
|
Maîtriser la peur : le cervelet, partenaire inattendu des troubles de lâhumeur |
|
|
| |
|
| |

Maîtriser la peur : le cervelet, partenaire inattendu des troubles de l’humeur
14 AVR 2023 | PAR INSERM (SALLE DE PRESSE) | BIOLOGIE CELLULAIRE, DÉVELOPPEMENT ET ÉVOLUTION | NEUROSCIENCES, SCIENCES COGNITIVES, NEUROLOGIE, PSYCHIATRIE
Les Axones du cervelet (en bleu) se retrouvent autour de neurones du thalamus qui vont vers le cortex préfrontal (vert) mais pas autour de ceux qui vont vers l’amygdale (rouge), autre région impliquée dans le contrôle des émotions. ©Equipe Neurophysiologie des Circuits Cérébraux, Inserm/CNRS/ENS-PSL
Les travaux d’une équipe pilotée par Clément Léna et Daniela Popa, directeurs de recherche Inserm à l’Institut de Biologie de l’ENS, parus dans Nature Communications révèlent l’importance du cervelet dans les émotions. Cette région du cerveau, surtout connue pour son rôle dans le contrôle moteur, agit aussi sur une aire du cortex préfrontal impliquée dans les émotions, et par ce biais régule l’extinction des souvenirs aversifs. Ce travail ouvre la voie à une meilleure compréhension de la régulation des émotions dans les troubles de l’humeur, mais aussi dans diverses conditions pathologiques comme par exemple l’anoxie périnatale, les tumeurs cérébrales ou encore les effets de l’alcool.
Pour survivre ou simplement optimiser son existence, mieux vaut être capable de reconnaître les risques et ajuster sa conduite en conséquence. Par des mécanismes associatifs, le cerveau apprend à identifier les indices annonciateurs du danger. Ces associations doivent être continuellement mises à jour, notamment pour reconnaître l’innocuité d’indices perçus jusque-là comme menaçants mais ne s’avérant plus associés à un danger.
Un défaut dans les processus de neutralisation de tels indices exposerait à accumuler seulement les associations négatives en conduisant, par exemple, à maintenir indéfiniment des réponses émotionnelles intenses à un traumatisme passé. Cependant, plutôt que d’oublier ces indices devenus négligeables, une des méthodes employées par le cerveau est de confier au cortex préfrontal le soin de réprimer leur signification aversive, grâce à un véritable apprentissage de l’innocuité, aussi appelé extinction.
En étudiant le cerveau des souris, l’équipe composée de chercheurs et chercheuses de l’ENS-PSL et de l’Inserm a montré que la zone du cortex préfrontal en charge de cette fonction reçoit des informations en provenance du cervelet (via un relai dans le thalamus, cf. figure). Les chercheurs sont parvenus à effectuer une inactivation ciblée de cette projection en y introduisant des récepteurs inhibiteurs artificiels. Ils ont observé que lorsque ce circuit est inactivé, la réponse de peur à un stimulus qui ne représente plus un danger se prolonge anormalement, ce qui indique un déficit d’apprentissage.
Les enregistrements de l’activité dans ce circuit cérébral ont alors révélé que le cervelet participe à l’apprentissage de la répression des souvenirs négatifs en stoppant des activités rythmiques cérébrales associées à l’état de peur.
Ces travaux complètent une étude précédente de la même équipe, qui avait démontré que l’intensité de l’association « indices – dangers » était également sous contrôle du cervelet, par des projections vers une autre région du cerveau que le cortex préfrontal.
Alors que le cervelet est très connu pour son rôle dans le système moteur (les effets moteurs de l’alcool sont par exemple à peu près entièrement dus à un effet sur le cervelet), ces démonstrations d’une régulation par le cervelet de la formation et de l’extinction de la mémoire émotionnelle sont importantes. Elles montrent que cette structure agit sur les processus clés de la régulation des émotions générées par nos expériences passées. Ce travail ouvre notamment la voie à une meilleure compréhension de la régulation des émotions dans les troubles de l’humeur, mais aussi dans diverses conditions pathologiques, liées par exemple à la sensibilité du cervelet à l’anoxie périnatale, aux tumeurs cérébrales ou aux effets de l’alcool.
DOCUMENT inserm LIEN |
| |
|
| |
|
|
|
Découverte du rôle dâun régulateur cérébral impliqué dans des maladies psychiatriques |
|
|
| |
|
| |

Découverte du rôle d’un régulateur cérébral impliqué dans des maladies psychiatriques
11 DÉC 2023 | PAR INSERM (SALLE DE PRESSE) | BIOLOGIE CELLULAIRE, DÉVELOPPEMENT ET ÉVOLUTION | NEUROSCIENCES, SCIENCES COGNITIVES, NEUROLOGIE, PSYCHIATRIE
Il était communément admis que des familles de récepteurs synaptiques transmettaient des messages excitateurs et d’autres inhibiteurs vis-à-vis des neurones. © Adobe Stock
Dans le cerveau, un récepteur supposément excitateur appelé GluD1 se révèle contre tout attente jouer un rôle majeur dans le contrôle de l’inhibition des neurones. Des altérations dans le gène GluD1 étant retrouvées dans un certain nombre de troubles neurodéveloppementaux et de maladies psychiatriques comme les troubles du spectre autistique (TSA) ou la schizophrénie, cette découverte ouvre la voie à de nouvelles pistes thérapeutiques pour lutter contre les déséquilibres entre transmissions neuronales excitatrices et inhibitrices associés à ces maladies. Ce travail, publié dans Science, est le fruit de collaborations de chercheurs et chercheuses de l’Inserm, du CNRS et de l’ENS au sein de l’Institut de biologie de l’ENS (IBENS, Paris) avec le laboratoire de Biologie moléculaire du MRC à Cambridge au Royaume-Uni.
La complexité du fonctionnement du cerveau révèle bien des surprises. Alors qu’il était communément admis que dans l’activité cérébrale, des familles de récepteurs synaptiques (situés à l’extrémité d’un neurone) transmettaient des messages excitateurs et d’autres inhibiteurs vis-à-vis des neurones, une étude copilotée par les chercheurs Inserm Pierre Paoletti et Laetitia Mony à l’Institut de Biologie de l’ENS rebat les cartes.
Pour bien comprendre de quoi il retourne, revenons aux fondamentaux. Une synapse « excitatrice » déclenche la création d’un message nerveux sous forme de courant électrique si un récepteur à sa surface peut se fixer à un neurotransmetteur excitateur présent dans l’espace interneuronal, le plus souvent du glutamate. On parle d’excitation neuronale. Une synapse « inhibitrice » empêche au contraire cette excitation neuronale en libérant un neurotransmetteur inhibiteur, souvent le GABA. On parle d’« inhibition neuronale ». Ainsi, la famille de récepteurs à glutamate (iGluR) et celle des récepteurs à GABA (GABAAR) ont a priori des rôles opposés.
Toutefois, un sous-type de récepteur au glutamate appelé GluD1 intriguait les scientifiques. En effet, alors qu’il est censé avoir un rôle excitateur, celui-ci est préférentiellement retrouvé au niveau de synapses inhibitrices. Cette observation, effectuée par l’équipe de la chercheuse Inserm Cécile Charrier à l’Institut de Biologie de l’ENS en 2019, avait interpellé la communauté scientifique car le gène GluD1 est souvent associé à des troubles du neurodéveloppement comme l’autisme ou à des maladies psychiatriques de type troubles bipolaires ou schizophrénie, dans les études génétiques de population humaine. Comprendre le rôle de ce récepteur représente donc un enjeu de taille. Pour y voir plus clair, l’équipe de Pierre Paoletti a étudié ses propriétés moléculaires et sa fonction, à partir de cerveaux de souris, au niveau de l’hippocampe où il est fortement exprimé.
Un rôle atypique
Les chercheurs savaient déjà que contrairement à son nom, le récepteur GluD1 ne peut pas se lier au glutamate. Mais dans cette étude, ils ont eu la surprise de constater qu’il fixait le GABA. L’équipe de Radu Aricescu à Cambridge a même décrit dans la publication la structure atomique fine du site d’interaction de GluD1 avec le GABA, grâce à une technique appelée cristallographie aux rayons X[1].
Son rôle dans le cerveau n’est donc a priori pas excitateur de l’activité neuronale mais inhibiteur. En prenant en compte ce résultat, peut-on toujours dire qu’il s’agit d’un récepteur appartenant à la famille des récepteurs à glutamate ?
« C’est en effet une interrogation mais toutes les analyses de phylogénie (les liens de parenté entre gènes et protéines) et les données structurales montrent qu’il en fait bien partie. En revanche, il est possible que certaines mutations acquises au cours de l’évolution aient profondément modifié ses propriétés fonctionnelles », explique Pierre Paoletti.
Autre curiosité, ce récepteur ne fonctionne ni comme un récepteur « classique » du glutamate ni comme un récepteur du GABA. Les deux provoquent en effet l’ouverture de canaux dans la membrane cellulaire permettant le passage d’ions responsables de l’excitation ou de l’inhibition du neurone. Le récepteur GluD1, lui, ne permet l’ouverture d’aucun canal. Son activité résulte d’autres mécanismes internes à la cellule qui restent à clarifier.
Enfin, ce travail suggère un rôle régulateur majeur de GluD1 vis-à-vis des synapses inhibitrices. En effet, lorsqu’il est activé par la présence de GABA, la synapse inhibitrice voit son efficacité augmenter. Cela se traduit par une réponse inhibitrice plus importante qui perdure pendant des dizaines de minute.
« Autrement dit, GluD1 renforce le signal d’inhibition. Peut-être en favorisant le recrutement de nouveaux récepteurs GABA à la synapse ? On peut en tout cas parler de régulateur clé », explique Laetitia Mony.
Pour les scientifiques ayant contribué à ce travail, cette découverte marque une véritable avancée.
« Ces résultats ouvrent la voie à une meilleure compréhension des déséquilibres entre messages excitateurs et inhibiteurs dans le cerveau en cas de troubles neurodéveloppementaux et de maladies psychiatriques comme les TSA ou encore la schizophrénie, ou dans des maladies comme l’épilepsie caractérisée par une hyper excitabilité neuronale. Dans un second temps, il sera important d’étudier si GluD1 peut constituer une cible thérapeutique intéressante pour rétablir un meilleur équilibre et réduire les symptômes dans ces maladies », concluent-ils.
[1] Il s’agit d’une technique d’analyse physicochimique qui se fonde sur la diffraction des rayons X par la matière pour connaître sa composition moléculaire et sa structure en 3D.
DOCUMENT inserm LIEN |
| |
|
| |
|
|
|
Une nouvelle technique dâIRM permet de localiser des foyers de cellules tumorales agressives et pourrait améliorer le traitement des glioblastomes |
|
|
| |
|
| |

Une nouvelle technique d’IRM permet de localiser des foyers de cellules tumorales agressives et pourrait améliorer le traitement des glioblastomes
13 NOV 2023 | PAR INSERM (SALLE DE PRESSE) | CANCER
Les glioblastomes sont des tumeurs cérébrales très agressives dont le traitement consiste en une chirurgie et une radiochimiothérapie. Une nouvelle technique d’imagerie médicale pourrait améliorer le pronostic des patients, selon un récent essai clinique mené par Élisabeth Moyal, professeure à l’université Toulouse III – Paul Sabatier et cheffe du département de radiothérapie à l’IUCT-Oncopole. Les résultats de son essai clinique mené à l’Oncopole et au sein de son équipe de recherche Inserm au Centre de recherches en cancérologie de Toulouse (CRCT – Inserm/CNRS/UT3), en collaboration avec le service de neurochirurgie du CHU de Toulouse, ont été publiés dans la revue Science Advances le 3 novembre.
Le traitement des glioblastomes consiste en une résection chirurgicale de la zone tumorale centrale (dénommée CE), suivie d’une radiochimiothérapie sur cette même région, ainsi que sur la large zone péritumorale infiltrée par les cellules tumorales qui y ont migré (appelée zone FLAIR). Malgré ce traitement, la plupart des patients vont présenter une rechute, notamment au niveau de ces régions péritumorales FLAIR qui n’ont pas été retirées lors de la chirurgie initiale.
Ce sont ces zones à risque qui ont été étudiées par la Pr Elisabeth Moyal et son équipe Inserm. En effet, en utilisant une technique d’IRM, appelée spectroscopie de résonance magnétique et qui permet d’analyser le métabolisme des tumeurs, l’équipe avait précédemment montré que les régions tumorales CE et péritumorales FLAIR affichant un hypermétabolisme prédisaient l’endroit où la tumeur allait récidiver après le traitement.
Les récidives sont principalement imputées à une sous-population de cellules tumorales plus agressives, les cellules souches de glioblastome. L’hypothèse des chercheurs a donc été que les régions hypermétaboliques étaient enrichies en cette population de cellules souches cancéreuses.
Pour la vérifier, un essai clinique a été mis en place chez 16 patients porteurs de glioblastome, bénéficiant en préopératoire d’une IRM classique associée à une spectroscopie de résonance magnétique permettant lors de la chirurgie guidée par ces techniques d’IRM de prélever des biopsies dans les zones tumorales CE et FLAIR, hypermétaboliques ou non. Les patients ont été ensuite traités par radiochimiothérapie standard.
Les expérimentations menées au CRCT par le Dr Anthony Lemarié, maître de conférences UT3, et Caroline Delmas, ingénieure de laboratoire à l’IUCT et au CRCT, ont confirmé leur hypothèse et ont démontré que les zones hypermétaboliques dans les régions péritumorales FLAIR étaient enrichies en cellules souches de glioblastome et surexprimaient de nombreux gènes impliqués dans l’agressivité et la résistance tumorale.
Plus les patients présentaient un fort enrichissement en cellules souches de glioblastome dans ces zones péritumorales hypermétaboliques, plus leur pronostic de rechute s’avérait sévère.
« La combinaison des deux techniques d’imagerie IRM et spectroscopie de résonance magnétique, avant la chirurgie, pourrait permettre une meilleure résection ainsi qu’un meilleur ciblage thérapeutique des zones hypermétaboliques présentes dans les régions péritumorales », précise la Pr Elisabeth Moyal.
Ce ciblage spécifique pourrait permettre d’améliorer le pronostic de ces tumeurs cérébrales très agressives.
DOCUMENT inserm LIEN |
| |
|
| |
|
| Page : [ 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 ] Précédente - Suivante |
|
|
| |
|
| |
|